
FDA’s Real-Time Clinical Trials Pilot Could Transform Drug Development
The approach is promising. The challenge is scaling it up.

Last week saw FDA unveil its real-time clinical trials pilot - a program designed to allow FDA to see clinical trial data in real-time, as it is being collected. The announcement was delivered with lots of excitement and fanfare: We saw FDA leaders on stage talking about re-engineering clinical trials, enabling them with AI, and reducing lag time. It felt like something exciting—perhaps even revolutionary—was happening.
Yet at the same time, I suspect that for the typical viewer it was hard to tell what that exciting revolutionary thing actually was. The headline made it seem simple: The FDA was piloting “Real-Time Clinical Trials… that will report endpoints and data signals to the agency in real time.” Today, FDA sees trial results only after the drug companies who run the trials, called sponsors, have collected the data, cleaned it, analyzed it, summarized it, and packaged it for review. This new approach could allow the agency to receive useful information earlier, reducing the amount of “dead time” between study phases.
But if you watched the FDA’s press conference, you might start to think that this announcement was about something else entirely. During the event, the pilot participants each shared some remarks. Most of them did not focus on real-time submission of data to FDA. Instead, they spoke about other things: a representative from Amgen talked about pragmatic trials. A researcher at UPenn talked about how the new pilot would ease the burden on their staff. Kent Thoelke, CEO of Paradigm Health, the technology developer for the pilot, spoke about the importance of bringing trials into community and rural settings. This is all great stuff. But what does it have to do with real-time submission of data to the FDA?
But there is a good reason that the pilot participants were speaking in such sweeping terms. The benefits of real-time trials could go far beyond just faster submission to the FDA. Real-time clinical trials represent a new, more efficient way of running trials that affects everybody involved—including study site researchers and patients. Beyond just reducing “dead time”, they hold the promise of making trials faster, less expensive, and more accessible to patients. In this post, I’ll explain why this program could change how trials are run, and what it will take to make it successful.
How trials got re-engineered to allow for real-time data
To understand why FDA’s pilot is potentially transformative, it’s helpful to understand how trials are run today, and how the real-time trials pilot changes that. As long-time readers of this blog already know, today’s trials are deeply inefficient and labor-intensive, largely owing to the cumbersome way in which we collect and clean trial data. Today, trial data is collected from a broad range of different “source” systems; EHRs, lab systems, and—far too often—on paper. Then, traditionally, those data are manually transcribed into an electronic data capture system (EDC): the system that is used to collate and transmit the trial data to the drug company. This manual transcription is time-consuming, error-prone, and can introduce lag between when data is first captured and when it’s submitted.
After the drug company receives the data from the EDC, it undergoes multiple layers of quality checks, which add more work and introduce even more delay. The drug company reviews the data for discrepancies and missing values, and if it finds an issue it will return a “query” to the study site. The site will then need to go back to the source documentation to resolve the issue. Then, there’s an even more labor-intensive step: A study monitor, hired by the sponsor, will fly out to the study site and review all of the data captured in the EDC, comparing it against the source data.
Meanwhile, the trial is collecting adverse events and endpoints. Each of these undergo extensive manual review. Adverse events are reviewed by the sponsor to determine whether they are serious, unexpected, and plausibly associated with the study drug. If so, they are shared with the FDA immediately. Endpoints are often reviewed by expert adjudicators, who confirm that the study endpoint was actually reached.
This is why real-time submission of data to the FDA is difficult. In the current model, each piece of data needs to be transcribed, validated, verified, and potentially adjudicated before it can be analyzed. The process can be long and drawn out. No sponsor or site would want to share “raw” data with FDA before this process was completed. This process is also a big part of the reason that we see so much dead time between phases. After the last patient visits a study site, there is still much work to be done: unaddressed queries, source data verifications, and adjudication. Only after that work is done for every data element will the sponsor sign off on a “database lock”, at which point the actual analysis of the trial data begins.
The data bottleneck begets a decision bottleneck: between study phases, drug companies must review their compiled data and decide what they want to do next. So after the database lock, the sponsor will analyze the data and make their critical go/no-go decision. They’ve probably already given some thought to how they will design their next phase, but after looking at the data they’ll confirm their design choices and share them with the FDA. They often will need to schedule a meeting with the FDA to share their proposed approach and make any modifications the FDA recommends (these interactions are especially common before phase III begins). Finally the drug company will start the long and laborious process of recruiting sites and patients for the next phase. Today, each of these steps is done sequentially, creating costly delays between phases.
How the RTCT pilot changes things
But what if we could do away with many of those steps? What if, instead of data being entered into disparate systems, the source data was entered electronically, then compiled and automatically imported into the sponsor’s electronic data capture system (or just sent directly to the sponsor)? Then, much of the manual work of data transcription, checking, and verification could be automated. And thanks to AI, we can even automate the downstream work: After the data is submitted to the sponsor and validated, AI could help classify the seriousness of adverse events and even adjudicate the endpoints. Data that previously took weeks to enter, clean, and validate can be available nearly instantly to analyze—and, of course, to share with the FDA.
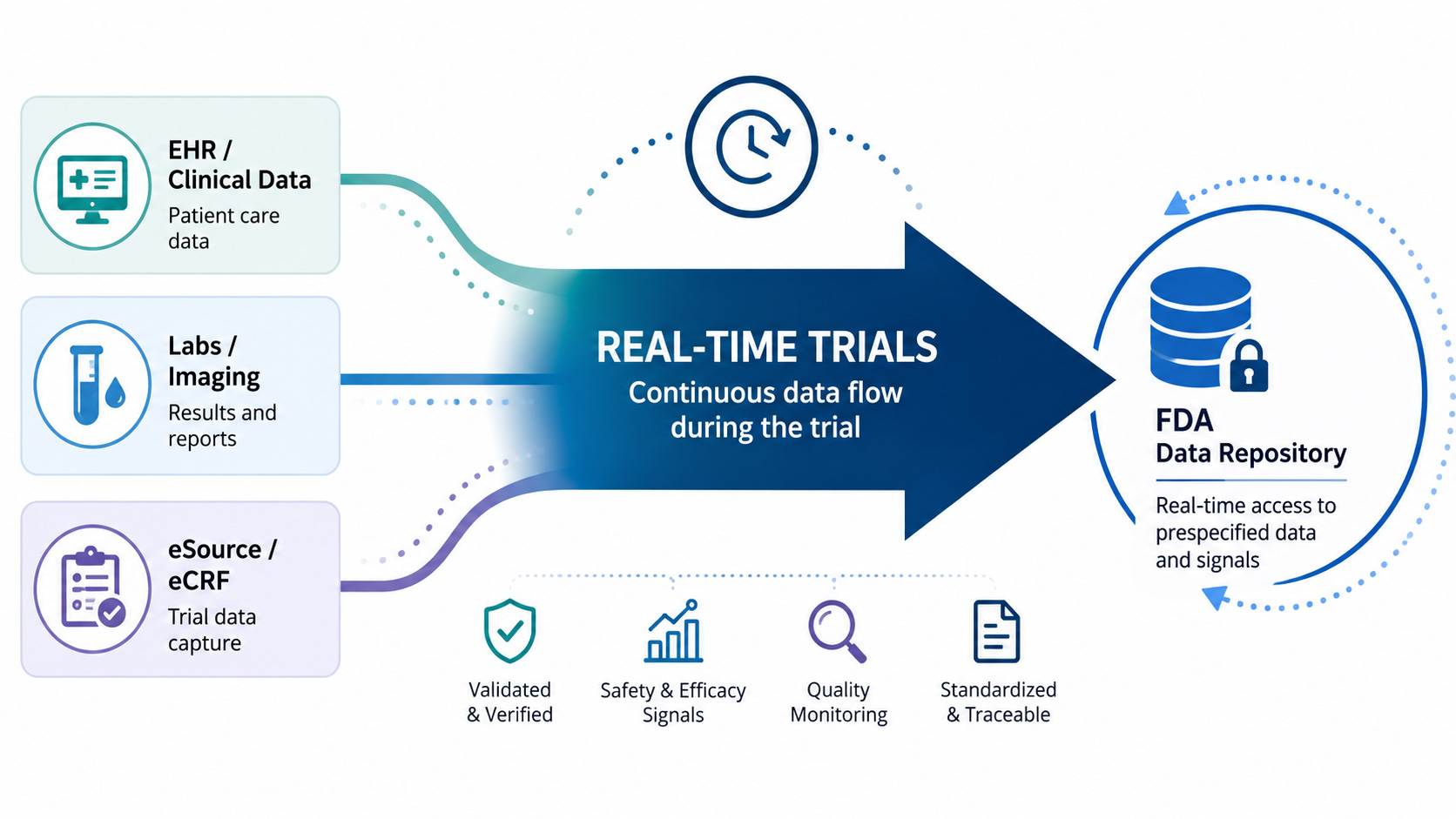
While I can’t claim to know precisely what Paradigm Health’s software is doing in these pilots, I suspect it is automating many of these tasks. And those kinds of automations yield many benefits beyond just enabling real-time submission to the FDA. We heard about many of them at the press conference. First off, the approach integrates data from disparate sources, reducing manual transcription work and errors. Sites will spend far less time dealing with source data verification and time-consuming queries. This saves an enormous amount of time and effort. Source data verification alone is believed to consume 25-40% of the trial budget.
This explains why FDA’s press conference kept drifting into the topic of site burden, community access, and pragmatic trials. The same infrastructure that makes real-time clinical trials possible also makes it easier for sites that don’t traditionally do clinical trials—like community and rural sites—to involve their patients in research. With much of the work automated, participation is simpler, especially when the trials themselves are designed to make use of the data collected in routine care.
Finally, when all of the systems that collect and use trial data are integrated and automated, it opens the door for even more innovation. Rapid, automated data analysis could make it easier to run adaptive and Bayesian trials by providing quicker feedback to sponsors about when a key milestone or threshold was hit. And the infrastructure that makes rapid data analysis possible also could be extended to automate many other tedious trial execution tasks, like patient recruitment, scheduling, and follow-up.
Will This Scale?
To be clear, the idea of using AI and automations to streamline trial data collection isn’t new; companies have been working on this for years. But FDA’s involvement could have a huge impact in actually getting these approaches adopted in a very conservative and risk-averse industry.
Observers are already asking how we can scale up this approach and apply it to more trials. Sean Khozin, who originated the real-time trials idea during his tenure at FDA, posted about this in his Substack: “The hard work, the transformation, is the scale-up. It is the second sponsor, and the tenth, and the hundredth. It is the standards work that has to happen quietly behind the announcements, the EHR-to-EDC plumbing, the sponsor data systems that have to be re-architected, the reviewer workflows that have to be redesigned around streams instead of submissions.”
If you are a longtime reader of my blog, you already know why scaling innovations in trials is so difficult. The trial industry is deeply fragmented, poorly coordinated, and risk-averse. And regulatory uncertainty makes companies fearful of doing things differently. You might also know that the industry has a long and inglorious history of piloting better ways of doing things, only to go back to doing things the old way after the pilot has ended.
The limits of this particular pilot illustrate the challenges. For the pilot, they picked a relatively simple task: the trials appear to be using one software vendor in just two health systems (this might change, but I expect the final number of vendors and sites to remain small). For this approach to be truly transformative, though, it will need to be scaled up to a much larger set of trials, including the large, expensive late-phase trials. These trials are conducted across multiple sites, often in multiple countries, using software from many vendors and pulling data from multiple EHRs and hospital systems. I don’t want to understate the technical achievement that Paradigm Health pulled off in developing the system used in this pilot—it’s difficult and impressive work. But expanding this approach will require a lot more technical work, including, as Sean Khozin noted, standards and plumbing.
FDA will need to be actively involved in this scale-up. Part of the reason that we do things in the current, inefficient way is because sponsors believe that FDA expects it. The cumbersome system of audits, source-data verification, and adjudications were not an inevitable part of trials; they grew over time to meet the perceived demands of regulators. If we want to automate those tasks with AI, sponsors will need to work closely with regulators to “de-risk” this approach. Pilots alone won’t be sufficient; FDA will need to set expectations, change policies, and even change its culture to accommodate a new approach to running and reviewing trials.
What Should the FDA Do Next?
As excited as I am about the potential of this new approach to running trials, the initial rollout was not exactly confidence inspiring. The FDA didn’t really tell a coherent story about why real-time trials matter, how the agency intends to use the data, or what it will take to scale up this work. But there’s still plenty of time for the FDA to provide more clarity—and I suspect they’re busy working on addressing these questions.
With that in mind, I’d offer three recommendations for the FDA:
First, the FDA needs to define the operating model for its use of real-time trial data. If you just read the press releases and materials, you might expect FDA reviewers to be monitoring a stream of data like a manager might monitor their corporate dashboard. Perhaps FDA reviewers will wake up in the morning and watch the trial results roll in while they drink their coffee. This, to be clear, would be a terrible idea. The last thing that sponsors need is FDA staring over their shoulders as they run the trial, (over)reacting to safety signals as they come in or prematurely drawing conclusions from interim views of study endpoints.
A more prudent model might look something like the one used in FDA’s real-time oncology review program, where the availability of early readouts allows FDA to gain familiarity with the trial outcomes earlier and therefore issue decisions more quickly. For example, FDA might receive a draft readout of the trial results the very day of the last patient visit before the trial’s “database lock”. This would give FDA a better sense of what to expect when the sponsor comes in later with its more polished data package.
But that is just one model of many the FDA should consider. Along with the real-time trials announcement, FDA published a request for information seeking input on potential pilots of AI-enabled methods to optimize early-phase trials.1 This could be a great opportunity to explore how these newer operating models might work in practice and anchor them in established principles for trial design and statistical analysis.
Second, FDA needs to help build the standards and infrastructure to scale this approach beyond just one vendor and a few trial sites. Today, this work is only possible because one company, Paradigm, worked closely with sites to integrate their disparate data sources. That is useful for a proof of concept, but it is not a scalable model for the broader clinical trials enterprise. Large late-phase trials (and even many early phase trials) are conducted across many sites, often in multiple countries, using multiple EHRs, EDCs, labs, imaging vendors, clinical outcome assessment tools, and monitoring platforms. For real-time trials to work, they’ll need to operate on common standards that all of these disparate systems can use.
FDA could play a role in setting data standards for real-time trials. FDA should work with ONC and standards development organizations to establish standards for how clinical trial systems share and transmit clinical and operational data. These standards could both facilitate real-time submission of data and help build the infrastructure needed to make trials more efficient and automated. These standards should include the metadata needed to understand the data’s provenance, its role in the trial, and whether and how it was verified or adjudicated. This work could begin now, building on work already underway in this area by existing standards development organizations.
Third, FDA should set clear expectations for sponsors when it comes to data collection, verification, and validation. The pilot shows that we can use technology to make trials far more efficient by reducing or automating cumbersome practices like source data verification, queries, and manual adverse event review. But for this to be possible, FDA needs to clearly define the conditions under which it will accept automated or AI-assisted alternatives, and how it intends to inspect study sites that use these approaches without requiring them to maintain reams of paper documents and PDFs. Otherwise, companies will default to doing the traditional, “safe” approach to data collection, even when better approaches are possible.
FDA’s expectations-setting should go beyond ordinary guidance. As readers of this blog know, FDA has tried in the past to issue guidance offering alternatives to practices like 100% source data verification, only to find that the guidance was not specific enough to change industry practice. I’d suggest FDA get more specific, identifying standards, practices, and data submission formats that it would deem acceptable for purposes of real-time submission and review.
If the FDA can address these issues, real-time clinical trials could go beyond just pilots and become the foundation for a better, more efficient way of running trials. But if FDA does not clarify its operating model, help build standards, and set regulatory expectations, this could go the way of most other trial pilots, which have failed to make any lasting impact on how the industry operates.
FDA will need to be thoughtful in how it manages this pilot program. Many (perhaps most) of the important questions around the use of AI in early-phase trials are not best answered through operational pilots; rather, they are scientific questions more appropriately addressed through things like validation studies and qualification programs. The RFI implies this operational/scientific distinction but does not make it clear. So I hope the FDA clarifies this and takes advantage of the pilots to figure out how the operations for this program will work.
A couple of things make a real and lasting change of this sort difficult.
First of all, the agency would have to commit up front to accepting certain kinds of data as evidence of efficacy and safety, which it has historically been extremely reluctant to do. The answer to questions in the general vein of "will you accept this" has always seemed to be "bring us the data and we'll see", which of course means that after you've spent hundreds of millions of dollars collecting that data, you do absolutely everything to make it bulletproof (and that risk aversion is exactly the problem you've been talking about, Adam). This is a serious cultural/structural problem.
Second is that you need long-term stable leadership at the FDA. The political chaos happening right now, with rumors of the head being fired and experienced leaders leaving, is... not that. We can hope that this changes with the next presidential administration, but that's a few years away, and more aspirational than rational.
The Sean Khozin quote really lands "the second sponsor, and the tenth, and the hundredth." That's where every promising trial innovation goes quiet. The pilot is the easy part; it's a controlled environment with a willing vendor and cooperative sites. The messy reality of a multinational phase III trial pulling from five EHR systems across three countries is a completely different problem.
The point about sponsor risk-aversion is underappreciated too. A lot of the cumbersome infrastructure we're now trying to automate away wasn't mandated. It accumulated because no one wanted to be the first company to do less of something FDA might later decide it actually wanted. That cultural inertia is arguably harder to fix than the technical plumbing.
Coming from a food science background, I find the parallel to food safety data systems surprisingly direct same fragmentation, same reluctance to share real-time data upstream to regulators, same pattern of pilots that look great in controlled settings and stall at scale. The bottleneck is almost never the technology.
Your three recommendations are exactly right, especially the third. Without FDA being specific about what it will actually accept, sponsors will keep doing SDV the old way just to be safe. Guidance that's too general has essentially no effect on a risk-averse industry, we've seen that pattern repeat.
Really useful breakdown of something that got a lot of fanfare without a lot of explanation.