
Clinical trial reforms that once seemed radical
How randomized controlled trials, preregistration, and results reporting became standard practice.

In an earlier post, I argued that clinical trials should be more transparent, and that data on individual patients’ outcomes should be anonymized and made available to other researchers for further research.
It’s easy to see that proposal as an ambitious reform, and in some ways it is. But the history of medicine shows us that clinical trials have already undergone a series of transformations that once seemed equally bold. Over time, the way we test treatments has become more rigorous and standardized. Improvements in transparency are a continuation of this trend.
In this post, I’ll give a brief history of clinical trial reforms and why transparency is the natural next step.
The rise of randomized controlled trials
One of the most consequential rigor-enhancing changes in clinical trials was the rise of the randomized controlled trial. In 1962, the United States passed the Kefauver-Harris Amendments, which required that new drugs demonstrate efficacy through adequate and well-controlled investigations before receiving approval. In practice, this pushed regulators and manufacturers toward randomized controlled trials (RCTs). This was seen as a dramatic shift.
At the time, clinicians had to rely on scattered uncontrolled studies, controlled studies without randomization, and case reports of claims of miracle treatments. They’d see some patients improving and others worsening, with no clear way to estimate the average effect or detect harms that only become visible at scale.
The requirement for controlled trials might have raised costs and overturned established practice. But it addressed a real problem: if you want to understand the effectiveness and safety of a drug using observational, uncontrolled data, you’d face many challenges:
Regression to the mean: Many conditions bring people to the doctor when symptoms are at their worst, and some improvement would have happened anyway. Someone might seek treatment for severe back pain after a particularly bad week, only for the pain to ease as the flare-up subsides. The same pattern appears in mental health: a person may begin therapy during an especially intense depressive episode, then improve as the episode naturally wanes. If improvement follows treatment, it’s tempting to credit the intervention, even when part of the change reflects a return toward a person’s usual baseline.
External events: Outcomes can also shift because the world around patients changes. For example, asthma admissions may spike during a period of high air pollution and fall when air quality improves, a heatwave can increase dehydration and kidney stress. Even public health campaigns, changes in food availability, or a stressful economic downturn can influence sleep, diet, and cardiovascular risk. If a treatment is introduced during such periods, its apparent effects may partly reflect broader changes.
Selection into treatment: People who receive a treatment often differ in systematic ways from those who do not. Patients who opt for a new preventive medication might be more health conscious, adherent to medical advice, or able to afford regular care. Conversely, a specialist clinic may see the sickest patients, or certain drugs may only be prescribed at severe stages of a disease, making treatment look less effective than it truly is.
Blinding and concealment address a different source of bias: the expectations and behaviours of the people involved in a trial. Even after randomization, participants or researchers may figure out who received the active treatment because the drug looks, tastes, or smells different from the placebo, or because it produces noticeable side effects. When this happens, it can partly reverse the benefits of randomization: if participants or researchers can identify who received the active treatment, differences between groups may reflect expectations and behavioural changes rather than the drug alone, meaning the estimated effect could be larger or smaller1 than the true effect of the treatment itself. Concealment is a first step, preventing researchers from knowing which treatment someone will be assigned to before allocation. Blinding goes further, keeping both participants and researchers unaware of group assignments throughout the trial.2
Without careful design, these differences can wrongly appear to be treatment effects. But adjusting for these factors is difficult, and the flexibility can also open the door to questionable research practices: analysing data in selective ways, choosing favourable subgroups, or stopping analysis when results look promising.
Randomization helps because it creates groups that, at the outset, have roughly equal propensities for the outcomes being studied. Darren Dahly has a clear explanation of this logic. And like my friend Julia Rohrer, I think of randomisation as the closest thing epidemiology has to magic.
In my view, there’s no question that some treatments did have good evidence for them even before the rise of randomized controlled trials. In particular, researchers could notice when treatments were followed by large, otherwise unexplainable changes – like the elimination of a disease following vaccination, or reversals of diseases that usually progressed quickly or fatally, as was sometimes the case after treatment with drugs like antibiotics and insulin.
But in most cases, it would have been hard to identify effective drugs, especially if their benefits were more modest, or if their benefits took a long time to become visible.
It’s useful at this point to break down randomized controlled trials into their components: an experiment, with randomization, a controlled group, and often the practice of ‘blinding’ as well. Each of these components has had many precursors.
Take controlled groups as an example, which trace back – at least as far as I knew – to 1747, when James Lind compared treatments for scurvy aboard a Royal Navy ship and found citrus fruit dramatically effective. [After publishing this, Erin Braid pointed out that there were far earlier experiments with controlled groups, including in Ancient Greece; James Lind’s experiment is commonly named as the first one or one of the first ones.] I also found it interesting to learn that some 19th and 20th century controlled studies assigned patients to treatment in an alternate sequence, as they arrived to see a doctor, which is somewhat close to randomisation, but doesn’t involve blinding the researcher or participant to which treatment they received.
Blinding has an even more interesting history. It traces back to at least 1784, when King Louis XVI commissioned scientists including Benjamin Franklin and Antoine Lavoisier to investigate the claims of Franz Mesmer, who said he could cure illness through “animal magnetism” (later named ‘mesmerism’ after him). Franklin and Lavoisier designed an experiment in which they physically blindfolded subjects while performing the magnetic treatment; but crucially, they sometimes withheld the treatment without telling the subjects.
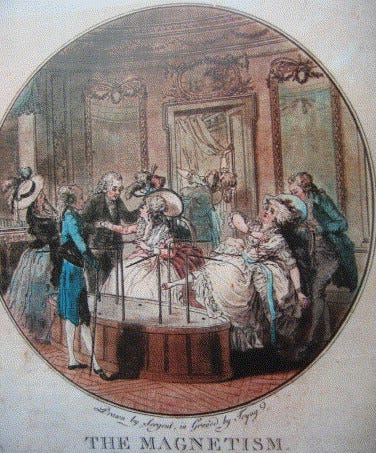
When they reported sensations regardless of whether they actually received the treatment, the experiment showed the effects were driven by their expectations rather than magnetism. It helped popularise the idea of blinding, in which patients are prevented from knowing which treatment they receive – although it was often the case that blinding was used to ‘debunk’ findings, rather than being open-minded to the results. Louis XVI’s interest, for example, was not purely scientific: mesmerism had become a fashionable and lucrative craze in Paris, and he likely worried about public disorder and the erosion of medical authority, giving the monarchy strong incentives to discredit it.
Combining these practices into randomised controlled trials, however, is relatively recent, at least historically speaking. The first widely recognised modern RCT was the UK Medical Research Council’s 1948 streptomycin trial for pulmonary tuberculosis.3 Patients were randomly assigned to receive the antibiotic streptomycin (a new breakthrough at the time) plus bed rest or bed rest alone. The trial showed clear improvements in survival and lung disease according to radiology scans in the treated group, but they also found early evidence of antibiotic resistance emerging.
As the twentieth century progressed, RCTs became more common. A very interesting example I’ve read of recently was the US National Institutes of Health’s Coronary Drug Project, conducted in the late 1960s and early 1970s, before statins were introduced.
In the decades leading up to it, researchers were experimenting with a surprisingly eclectic set of ways to lower cholesterol. If high cholesterol drove coronary heart disease, then lowering it ought to prevent heart attacks, so investigators tried hormones, vitamins, industrial resins, and metabolic drugs. Doctors claimed various drugs might be effective:
Thyroid hormone (notably dextro-thyroxine)
Oestrogens
Niacin (vitamin B3)
Bile-acid sequestrants such as cholestyramine
Fibrates
The Coronary Drug Project trial allowed for a proper, large-scale testing of these drugs in a randomized controlled trial to see whether they actually reduced heart attacks and mortality. The results were surprising – niacin and cholestyramine showed meaningful benefits, but some other treatments, including oestrogen therapy and dextrothyroxine, actually slightly increased mortality – and changed clinical practice.
By the 1970s, randomized controlled trials were widely accepted as a gold standard for evaluating therapies. And after the 1962 reforms in the United States, along with similar regulatory shifts elsewhere, controlled evidence became the norm for drug approval.
Preregistration
Randomization solved the design problem, but new challenges emerged in how results were analyzed. By the late twentieth century, the environment surrounding drug regulation had changed profoundly. To bring a medicine to market, companies were now expected to demonstrate both safety and effectiveness, usually through at least two well-controlled clinical trials, and typically using randomized controlled designs.
This was a major improvement over earlier eras, but it also raised the stakes dramatically. When development costs run into the hundreds of millions and a successful drug may earn billions annually, a failed trial can mean the loss of an enormous commercial opportunity.
The 1980s and 1990s saw a wave of breakthrough therapies reach patients. Statins transformed the prevention of cardiovascular disease. Angiotensin-converting enzyme (ACE) inhibitors improved survival in heart failure. Antiretroviral drugs began to turn HIV from a fatal diagnosis into a manageable condition.
But not every promising therapy proved effective, and even successful drugs often looked more impressive in early studies than they did later in routine practice. Understanding why requires a brief detour into how trial results are judged.
In most trials, success is judged using a statistical test that produces a p value, roughly the probability of seeing a result at least this extreme assuming the treatment has no real effect. By convention, results are called “statistically significant” if this probability falls below 0.05.
At the margins, this threshold creates a temptation. When a trial narrowly misses its endpoint, researchers may search for a significant result elsewhere: in a secondary outcome, a composite measure, or a subgroup analysis that, by chance, clears the bar. Given the enormous financial investment involved in developing a drug, there are strong incentives to tweak analyses at the margins, for instance, by changing which outcome is analyzed after seeing the data, a practice called outcome switching.
This flexibility undermines the validity of results. While randomisation improved how trials are designed, it did not fully address how results are analysed and reported; and flexibility after seeing the data creates opportunities for finding results that are favorable but unreliable.
To reduce the scope for this type of post hoc reinterpretation, regulators and journals began requiring trial protocols to be registered in advance. Starting in the late 1990s and expanding in the early 2000s, investigators were asked to document their study design and prespecified outcomes before data collection was complete. In the United States, these requirements broadened over time and were formalized through federal law and FDA regulations, with trial protocols and outcomes posted on ClinicalTrials.gov. Registration also became a prerequisite for publication in major journals, reinforcing the norm across the research community.
Analyses of trials funded by the National Heart, Lung, and Blood Institute found that before prespecification became standard, published trials were more likely to report positive results. After registration became mandatory, the pattern changed: many trials found no effect, and some detected harm. It became harder to redefine what success meant.
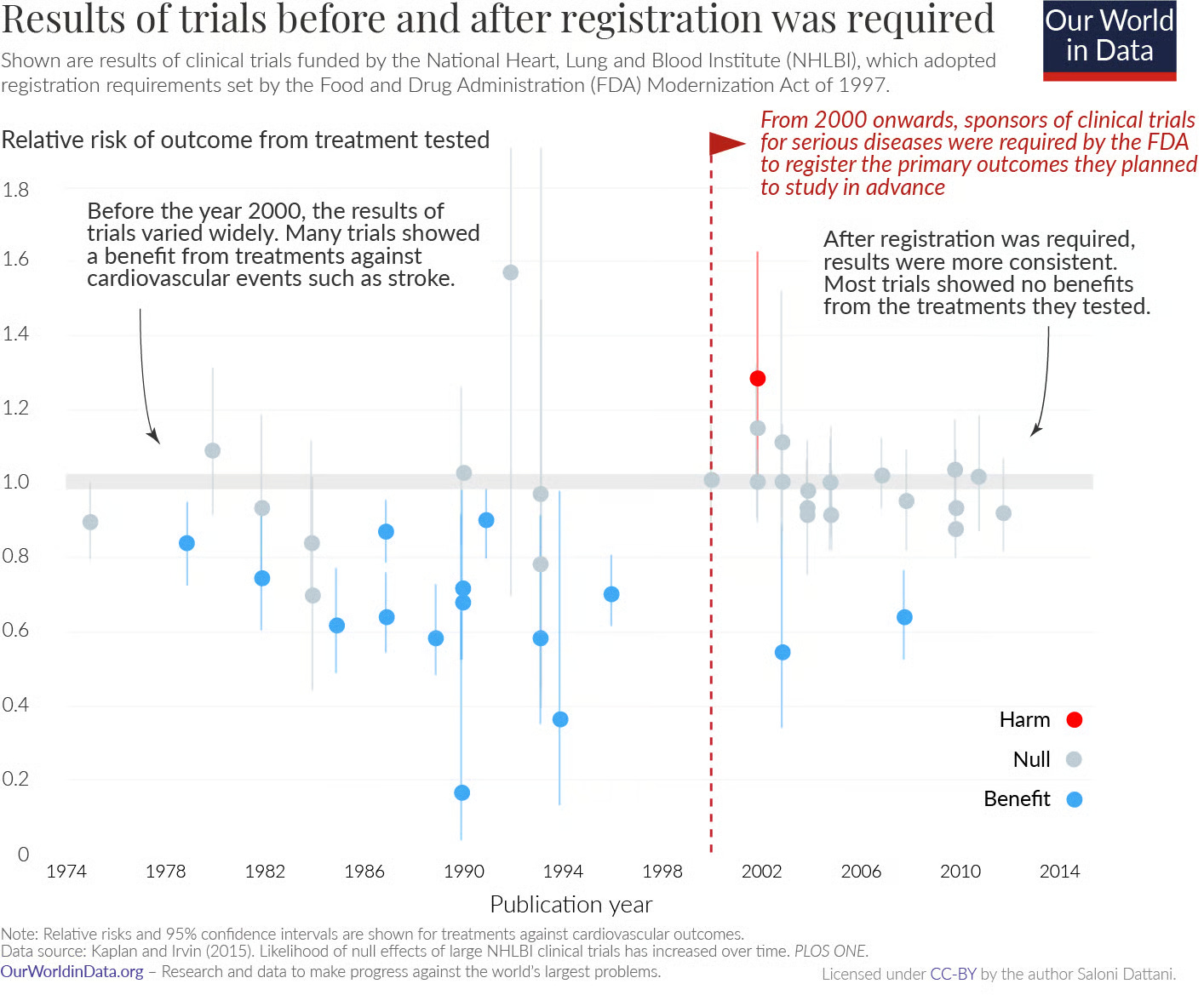
Even so, the value of prespecifying analyses is not about eliminating judgement but in improving transparency. It allows readers to see what researchers initially set out to measure, what changed, and why. There are countless examples from history of how both successes and failures in science have contributed to knowledge; science advances by testing predictions, learning from the results, and refining theories over time.
Trial reporting
By the late twentieth century, clinical trials had become the gatekeeper for drug approval. Regulators increasingly required convincing evidence of both safety and effectiveness, usually from multiple well-controlled studies, before a medicine could reach the market. This shift improved the reliability of medical evidence. But even as trial design grew more rigorous, a problem remained: what happened to the results once a study ended.
The results of many clinical trials were never published. Studies showing clear benefits were far more likely to appear in medical journals, while trials finding no effect or suggesting harm often remained unpublished. The problem became increasingly visible in the 2000s, as systematic reviewers began documenting “missing” trials.
Analyses of antidepressant trials comparing journal publications with the full set of studies submitted to regulators found many studies went unpublished, and were disproportionately those with negative or null results (see here for a more recent analysis, which finds improvement). The published literature suggested overwhelming effectiveness, while the complete dataset showed more modest benefits. When meta-analyses relied only on published studies, average effect sizes were inflated, meaning clinicians and patients were often seeing an overly optimistic picture of drug effectiveness.
There are probably many reasons for this: Journals prefer novel and positive findings, and often require multiple submissions and long delays before acceptance, companies have little commercial incentive to highlight disappointing results, academic researchers face career pressures that reward publishing and success, and negative results seem less actionable. The path of least resistance is to simply move on, hopefully to something more promising.
But the implications are not merely academic. Incomplete reporting makes it harder to learn from failures and adjust drug development plans, to estimate the true average benefit of treatments, to detect rare harms, and to make informed decisions about patient care.
Since the 2000s, governments and regulators began introducing legal requirements for trial registration and results reporting. The 2007 FDA Amendments Act in the United States required many trials to post summary results in a public registry. In Europe, transparency rules evolved into the EU Clinical Trials Regulation and a centralized database designed to make trial information publicly accessible. These policies aimed to ensure that results entered the public record regardless of whether they were favourable.
Compliance did not improve overnight and enforcement was initially limited, but pressure emerged from multiple directions. The AllTrials campaign pushed for the registration and reporting of all trials, the Good Pharma Scorecard tracked company transparency commitments, and the FDAAA TrialsTracker made reporting performance publicly visible. In the United Kingdom, parliamentary scrutiny and reviews by the National Audit Office and the House of Commons Public Accounts Committee criticized failures to report trial results, increasing political and public pressure.
This combination of regulation, monitoring, and public accountability gradually shifted behaviour. Reporting rates rose substantially in the United States after reporting requirements took effect. Recent research by Cunningham et al. finds that academically funded trials initially had very low compliance rates, but rose rapidly. Industry funded trials began at higher compliance rates, and also increased. The researchers also found that this shifted the behaviour of other firms, which became more likely to wait for trial results before advancing similar drug candidates. As with earlier reforms, mandatory reporting was initially framed as a bureaucratic burden, but it’s increasingly considered basic infrastructure for trustworthy medicine.
Today, roughly 75–80% of clinical trials that meet the FDA requirement report their results within a year, with higher rates of reporting among industry-funded trials than academic trials.
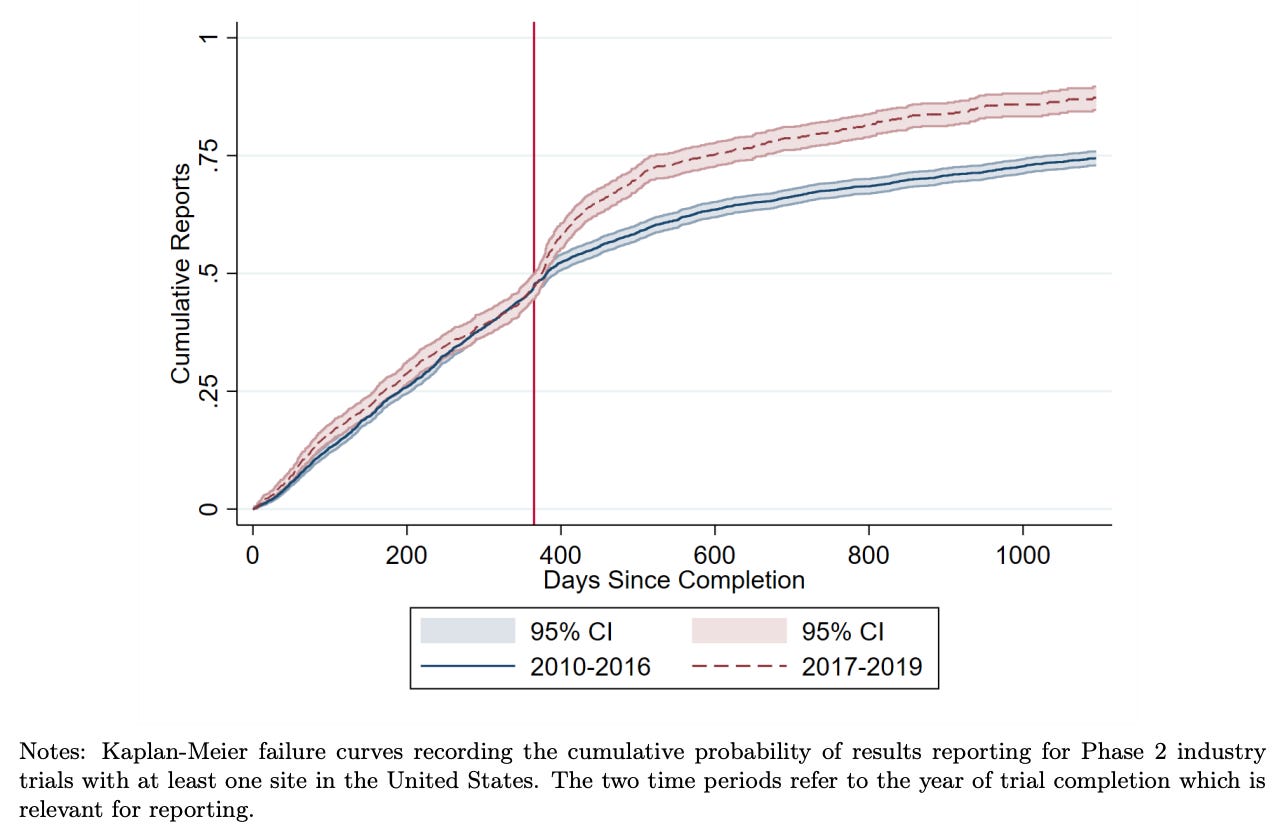
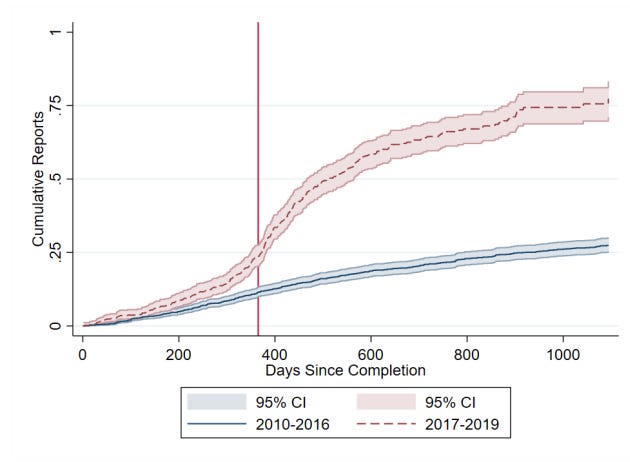
Conclusion
This blog is about clinical trial abundance, but it’s worth stepping back to ask why we care about trials at all. Clinical trials are not an end in themselves; they exist to generate reliable information about whether treatments are safe and effective. In that sense, it’s not just clinical trials that we want an abundance of, but information from them. The reforms described above, randomization, preregistration, mandatory reporting, addressed ways that information was being distorted or lost. Data sharing is the natural continuation of that arc.
In my previous post, I argued that sharing individual patient data could unlock a range of benefits that go beyond what published results alone can offer: pooling data across trials allows meta-analyses to detect effects on rare outcomes and confirm or rule out rare harms; it helps make sense of inconsistent results by letting researchers explore whether differences across trials reflect chance, patient populations, or study design; it makes it easier to ask new questions of old data, including when drugs developed for one condition show unexpected benefits for another; it enables better head-to-head comparisons between treatments, and can help tailor recommendations to patients’ characteristics; and it reduces redundancy: when trial results are visible, researchers can learn from failures and avoid repeating them.
So why isn’t this already standard practice? For industry-funded trials, the commercial disincentive is straightforward: sharing data helps competitors at least as much as it helps you. A firm that shares its patient data is effectively subsidising rivals’ research programs. But if all firms shared, the field as a whole would benefit, and so would each firm within it, gaining access to a much larger pool of evidence than any single company could generate alone. The case for coordination, whether through regulation or shared norms, follows from that.
For academic trials, the commercial disincentive largely disappears, and most academic research is publicly funded, which creates its own obligation to make results available. But it’s still uncommon to share data, probably as a result of practical frictions, career incentives that reward further publications foremost, and the absence of strong mandates. The relative neglect of data sharing in academic trials is harder to justify.
The case for sharing individual patient data from trials is strong enough that I think it warrants the same kind of coordinated push that succeeded for trial registration and results reporting. The answer could be better incentives, data infrastructure, and in some cases mandates from funders and regulators to make sharing practical and beneficial rather than burdensome.
Scientists often say that the best research doesn’t just answer a question, it opens up new ones. That’s how I think about data sharing as well. Without individual patient data, we’re left mainly with headlines, with limited ways to verify or explore the data to answer those questions, or build upon accumulated knowledge.
I’d like to thank Matt Clancy, Dylan Matthews, Nisha Austin, Ruben Arslan, Jamie Cummins, and Adam Kroetsch for feedback that improved this post.
Incomplete blinding can sometimes result in an underestimated effect size if participants who guess they received the active treatment change their behaviour in ways that work against it. For example, in trials of vaccines, participants who suspect they had been vaccinated may feel more protected and take fewer precautions against infection, increasing their actual exposure relative to the placebo group. This would make the vaccine appear less effective than it truly was.
It is sometimes impossible to blind participants to their treatment, such as for experiments involving talking therapies or when a safe or convincing placebo doesn’t exist, but researchers can instead focus on verifiable outcomes like blood markers or mortality that are less susceptible to expectation effects, though they may still be affected by behavioral changes.
While the streptomycin trial is famous, there were earlier, less-publicized RCTs. One example is given by Bothwell et al., who write: “in 1931, James Burns Amberson and colleagues published a study in which a coin flip randomly determined which of two seemingly equally divided groups of patients would receive sanocrysin for the treatment of tuberculosis.” (Here’s a link to that study)